在做 qPCR 时,你是怎么计算 cDNA 的加入体积的呢?小编也经常收到来自小哥哥小姐姐同样的疑问,那 cDNA 投入多少体积、稀释多少倍,如何去判定投入的量是否准确呢?接下来我们就一起来了解一下吧!

最常见的 2 种cDNA投入量的做法
实验室可能会有一套 SOP,只要按照师兄师姐告诉我们的 cDNA 稀释倍数进行稀释就可以了!
NO.2 使用 Nanodrop 测定 cDNA 的浓度
使用 Nanodrop 测定一下 cDNA 的浓度,按照以往的经验值进行添加 cDNA,或者按照师兄师姐的 SOP 上写到的浓度进行添加。
那这两种做法有什么弊端呢?
首先:不同处理组的 RNA 的表达量是不一样的,如果师兄师姐的 RNA 表达量比较高,例如稀释 1000 倍,CT 值为 25,而您的 RNA 表达量比较低,cDNA 原液投入 CT 值已达到 30,那如果您还按照 1000 倍进行稀释cDNA,那 CT 值已然超出 35 Cycles 了,即便出现扩增曲线,那也是无效的值。
备注:在 SYBR qPCR 中,CT 值超出 35,认为 CT 值是无效的。
如果不稀释,直接用 cDNA 原液做呢?又会面临高表达基因的 CT 值偏小,可能已超出 qPCR 线性范围之外,导致最终定量结果不准确。
其次:使用 Nanodrop 测定 cDNA 浓度是否可行呢?我们先一起了解一下 Nanodrop 测定核酸的原理吧!
核酸是由核糖、磷酸基以及碱基构成。其中,由于碱基含有芳香环结构,因此具有紫外吸收的特性。核酸吸收波长为 260 nm,根据朗伯比尔定律,已知物质的消光系数,液层的厚度,就能利用其吸光度来计算出物质的浓度。
与此同时,280 nm 处可检测蛋白质等;230 nm 处可检测盐离子、去污剂、苯酚、乙醇、糖类等,所以还能通过计算 A260/A280 和 A260/A230 的比值,估计核酸纯度(纯 DNA:A260/A280≈1.7-1.9;A260/A230≈2.0-2.2;纯RNA:A260/280≈2.0;A260/A230≈2.0-2.2)。
但紫外吸收法对溶液的 pH 和盐浓度极为敏感:吸光度本身会受到离子强度以及 pH 这两个因素的影响,所以容易受到杂质的干扰。未纯化的产物 Nanodrop 浓度测定结果会受到 RNA 残留、蛋白残留、盐离子、糖类等物质的影响,导致测定值虚高。
而我们拿到的 cDNA 其实就是一个未纯化的产物,里面有逆转录后残留下来的 Buffer、逆转录酶、引物核酸等,会严重干扰 Nanodrop 对 cDNA 浓度的测定。所以使用 Nanodrop 测定 cDNA 浓度的做法也是不准确的。
可能说到这里,有的小伙伴会有疑虑,既然 Nanodrop 测定未纯化的核酸的浓度会产生虚高的现象,那把 cDNA 纯化一下就 OK 啦?纯化是可以去除杂质,使 cDNA 的浓度测定趋于准确,但是纯化回收的效率并非 100%,定会损失掉一部分核酸,那必然会对后续定量的结果造成影响。
常见的这两种 cDNA 投入量的做法都不是很准确,那应该如何确定 cDNA 的稀释梯度呢?
如何准确确定cDNA的稀释梯度及投入量?
方法一
将cDNA原液做梯度稀释,把得到的不同梯度 cDNA 同时进行 qPCR,选择位于 15-33 范围内的 CT 值对应的 cDNA 稀释梯度作为最佳稀释梯度。如下图所示。(经验值认为,CT 值在 15-33 范围内是准确的)
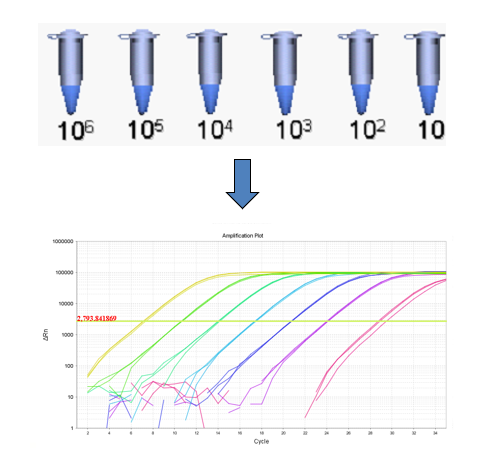
举个例子:如要扩增某处理组 GAPDH 内参基因,将逆转得到的 cDNA 进行 10 倍梯度稀释,得到的 cDNA 浓度梯度分别为:100(原液)、10-1、10-2、10-3、10-4,将 4 个 cDNA 模板同时上机做 qPCR,得到的 CT 值分别为:12、15.3、18.6、21.9,所以可以选择 cDNA 的最佳稀释梯度为 10-2-10-4,后续再扩增该处理组 GAPDH,就可以选择 10-2、10-3、10-4任何一个梯度。
方法二
若拿到一个之前没有做过的体系,不知道其拷贝数高低时,可以先选择 cDNA 原液进行 qPCR 并得到 CT 值。根据公式:2△CT=浓度差,进行调整最佳 cDNA 稀释梯度。
举个例子:某处理组 GAPDH 内参基因,cDNA 原液上样 1 μl,得到的 CT 值为 10。CT值=10,显然偏小了,要想使该处理组 GAPDH 内参基因的 CT 值为 20,应该如何计算稀释梯度呢?依据公式「2△CT=浓度差」可知,△CT=20(预期 CT 值)-10(实际 CT 值)=10,那浓度差=210=1024,也就是说在 cDNA 原液的基础上稀释 1024 倍即可使该处理组 GAPDH 内参基因的 CT 值达到 20。
不同的体系(基因)、同一基因不同的处理组都需要进行最佳 cDNA 稀释梯度的验证,保证所有的体系的 CT 值都落在 15-33 范围内,可以有效避免因 cDNA 稀释梯度不准确导致 CT 值过小或者 CT 值过大,甚至无 CT 值的情况。
注意:cDNA 原液的添加体积不能超出 qPCR 反应体系的 1/10,如 20 μl qPCR 反应体系,cDNA 原液最多只能上 2 μl,超出 1/10 体系,会对 qPCR 反应产生抑制作用。
有些小伙伴读到这里可能会有疑问了,如果内参基因表达量较高,使用 cDNA 原液 CT 值=11;目的基因表达量较低,使用 cDNA 原液 CT值=32,不管怎么稀释,好像都没有一个可以折中的 cDNA 稀释梯度,这种情况下该如何稀释 cDNA 呢?
有些小伙伴读到这里可能会有疑问了,如果内参基因表达量较高,使用 cDNA 原液 CT 值=11;目的基因表达量较低,使用 cDNA 原液 CT值=32,不管怎么稀释,好像都没有一个可以折中的 cDNA 稀释梯度,这种情况下该如何稀释 cDNA 呢?
极端情况下如何把控cDNA的稀释梯度?
最简单直接的方法就是:目的基因和内参基因分别进行稀释!目的基因减少稀释梯度甚至不稀释,内参基因增加稀释倍数。
接着上面例子,我们可以稀释 512 倍 cDNA 原液,以其为模板扩增内参基因,使 CT 值为 20 左右;继续使用 cDNA 原液扩增目的基因,保证其 CT 为 32。这样就解决掉无法同时兼容内参基因与目的基因的 cDNA 稀释倍数的问题啦!
但这样做的前提条件是:保证所有组的内参基因为相同的 cDNA 稀释梯度,所有组的目的基因为相同的 cDNA 稀释梯度。
为什么可以这么稀释呢?这种方法可行吗?会影响相对定量计算的结果吗?
我们先来回顾一下相对定量的计算公式:

该公式是处理组的△CT值-对照组的△CT值,而△CT=目的基因CT值-内参基因CT值。
① 当目的基因对应 cDNA 不稀释,内参基因对应 cDNA 稀释 512 倍,那目的基因与内参基因的差值(△CT)会存在一个稀释梯度不一致的差值;
② 当处理组的 △CT 值-对照组的 △CT 值时,这个稀释梯度不一致的差值就会被减掉;
③ 所以即便内参基因与目的基因对应的 cDNA 稀释梯度不一致,对计算结果仍然没有影响,稀释梯度不一致的差异最终是会被减掉的。但前提一定要保证我们上述提到的前提条件哦!
磨刀不误砍柴工,小伙伴们一定要确定好 cDNA 稀释梯度后再继续后续的 qPCR,这样才能事半功倍。关于 cDNA 如何稀释的方法,您是否已经掌握了呢?
明确了 cDNA 稀释梯度的方法确实可以帮助我们获得更加准确的实验数据,但是想要做好逆转录和定量实验,选好「武器装备」才是最为重要的。小 V 今天就给大家带来一对「黄金搭档」,在他们的帮助下,不仅可以在短时间内一步获得高质量的 cDNA,同时最终可以获得高平台期的优质定量曲线。话不多说,让我们一起来认识一下他们吧。
产品名称:HiScript III All-in-one RT SuperMix Perfect for qPCR
产品货号:R333
产品优势
15 min 一步完成基因组清除与逆转录;
cDNA 长久存放无降解:cDNA 可于 37 ℃ 存放 7 天、4 ℃ 存放 4 周、反复冻融 30 次, 依然具有良好的稳定性。
产品名称:Taq Pro Universal SYBR qPCR Master Mix
产品货号:Q712
产品优势
超高的扩增平台值:扩增产量大幅度提升,拥有超高扩增平台值
广泛的平台适用性:特殊 ROX 参比材料,适用于所有 qPCR 仪器,无需在不同的仪器上调整 ROX 浓度