
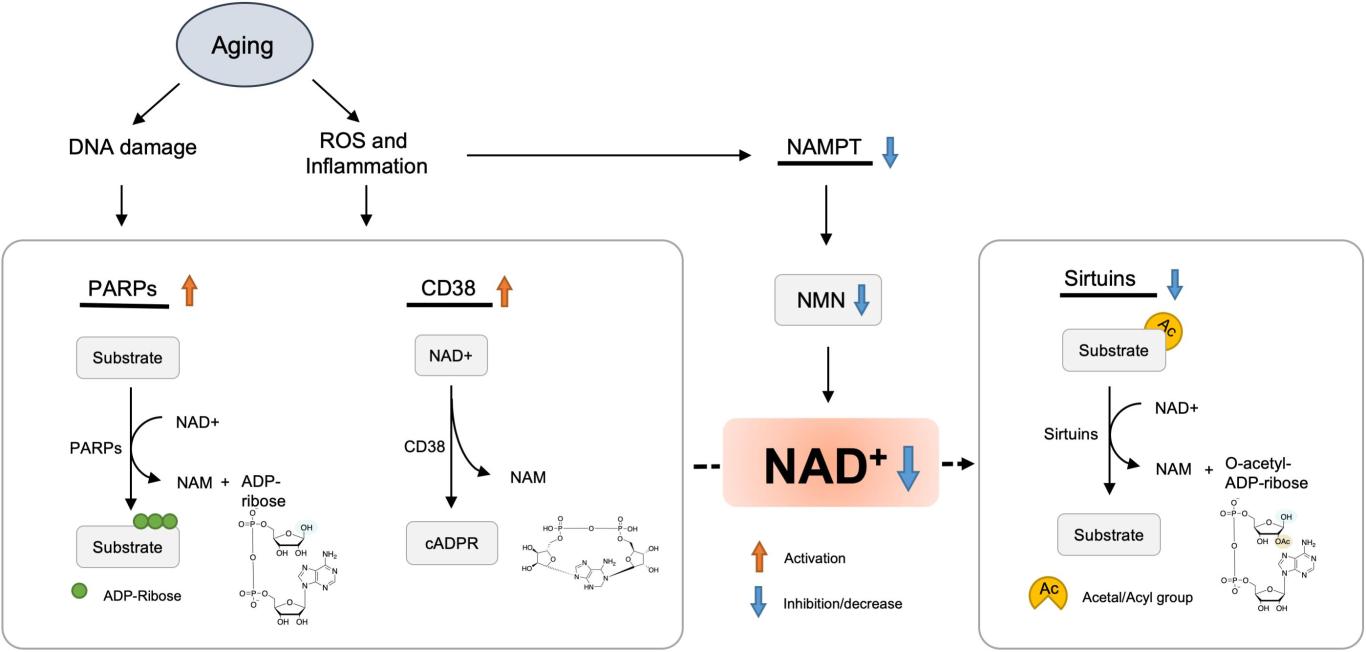
NAD+缺乏加速衰老
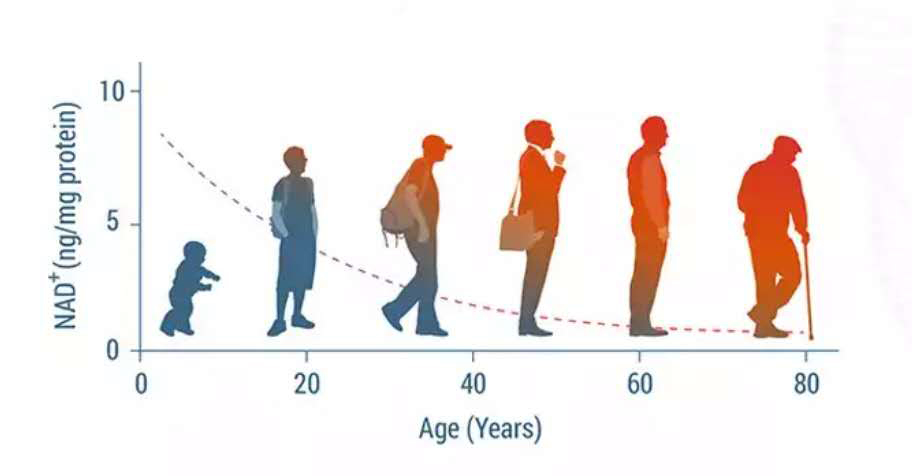
现在人们普遍认为,在衰老过程中,NAD+水平会逐渐下降,NAD+消耗酶的活性受到NAD下降的影响,如sirtuins和PARPs,它们通过调节p53、NF-κB、PGC-1α和HIF-1α等信号通路,导致氧化损伤、代谢紊乱、昼夜节律异常和线粒体功能障碍[1-4]。因此,增强NAD水平是改善寿命和抗衰的一种新思路。
NAD+缺乏会加速衰老。NAD+水平在老蠕虫中稳步下降,导致寿命进一步缩短。同样,小鼠和大鼠在衰老过程中出现了各种组织中的NAD+下降,如肌肉、脂肪组织、大脑、皮肤、肝脏和胰腺[5]。在老年人的大脑和肝脏中也可以观察到NAD+的减少[6]。与此相一致的是,血浆中NAD+及其代谢物NADP+和NAAD的水平在衰老过程中也显著下降[7]。
NAD+的下降是生物功能障碍和年龄相关病理进展的主要驱动因素。一些相关研究发现,NAD+的基因和药理学补充改善了与年龄相关的生物学功能,并增加了蠕虫和小鼠的寿命[8,9]。



NAD下降的一个重要原因是NAMPT介导的NAD生物合成的减少。在年龄诱导的T2D模型中,肝脏、骨骼肌、WAT和胰腺等各种组织中NAD+水平和NAMPT的表达受到严重抑制。NAMPT下降可能是由于慢性炎症和生物钟受损所致[10,11]。
NAD+随着年龄的增长而下降的另一种解释是PARP或CD38导致的NAD+消耗量增加。CD38的蛋白水平和酶活性在衰老过程中都有所增强,导致哺乳动物与年龄相关的NAD+下降。CD38还通过调节SIRT3的活性来导致线粒体功能障碍[12]。CD38的表达可能是由慢性炎症引起的,这是衰老过程中的一个特征之一[12]。PARP1活性增加,可能是由于DNA损伤的积累,这种持续的PARP激活进一步耗尽NAD池,并导致SIRT1活性降低。
一个关于衰老的著名理论认为,线粒体的衰变是衰老的关键驱动因素。线粒体功能障碍是衰老的标志,NAD+在维持线粒体功能中起着至关重要的作用,与衰老相关的NAD+下降会导致线粒体功能障碍。
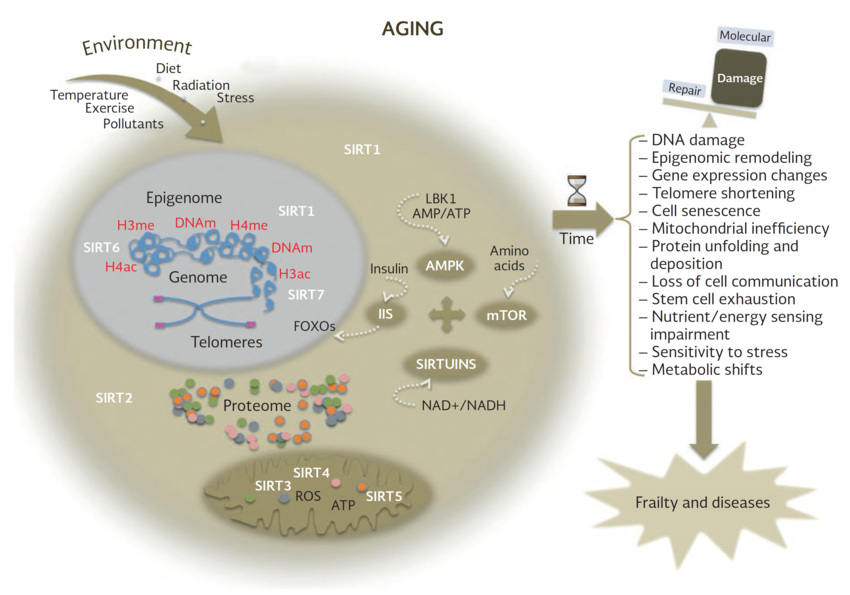
一组与抗衰老作用相关的蛋白质sirtuins,是依靠NAD+正常运作。Sirtuins在调节细胞和线粒体健康方面发挥作用。一些研究表明,Sirtuins有助于维持端粒酶的长度,而端粒酶的长度和寿命有关,sirtuin蛋白的激活有助于延长寿命。
越来越多的证据表明,NAD+依赖的SIR蛋白可以延长酵母、蠕虫、苍蝇和小鼠的寿命,并减轻许多与衰老相关的病理疾病。例如,大脑特异性或全身过表达sirt1的转基因小鼠都表现出了衰老速度减缓和寿命延长[8,13]。PARP抑制剂和NAD+前体能够调节NAD+可用性,通过SIR-2.1调节线粒体功能,延长寿命[8]。
老年小鼠肌肉和肝脏中的PAR化也显著增加,同时NAD+水平显著下降。由于CSB可以通过从受损DNA中置换PARP1来限制PARP1的活性,因此在CSB缺陷细胞和小鼠中,PAPRPs介导的PAR化增加,并占细胞NAD+消耗的大部分。PARP的这种异常激活抑制了SIRT1活性和线粒体功能障碍,这一过程可以通过PARP抑制剂和NAD+前体来挽救[14]。
衰老相关的核NAD+下降通过SIRT1-HIF-1α-c-Myc通路抑制线粒体编码基因,而提高NAD+水平则以SIRT1依赖的方式挽救老年小鼠的线粒体功能[15]。NAD+也通过改变SIRT3的活性来影响线粒体中氧化酶的乙酰化和活性[16]。
越来越多的人意识到,氧化损伤是与年龄相关的细胞功能恶化的一个重要驱动因素[17,18]。 老年人脑中的DNA氧化损伤和蛋白质氧化与抗氧化酶活性下降有关[19,20]。与年龄相关的氧化应激和细胞衰老的增加,会导致细胞更容易发生坏死性凋亡,从而释放DAMPs,触发炎症反应[21]。促炎细胞因子反过来增强线粒体和NOXs生成的ROS,促进氧化损伤的进一步积累[22-25]。
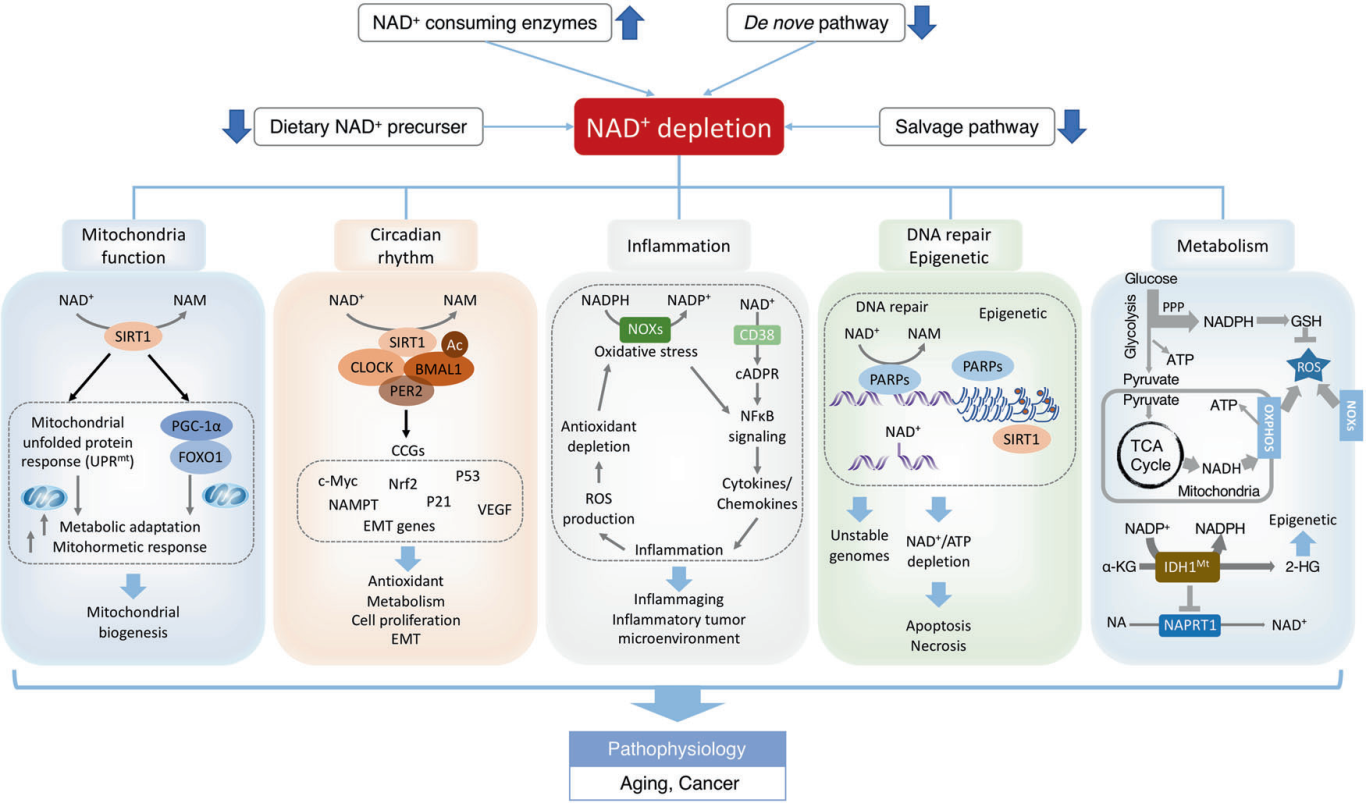
NAD+可改善衰老过程中的氧化损伤。NADH/NADPH是缓冲氧化应激的强大还原供体,保护细胞/组织在衰老过程中免受氧化应激。老年大鼠NAD+浓度和NAD+/NADH比值显著降低,同时氧化应激增强,抗氧化能力降低。在离体主动脉中添加NMN可提高NAD+和MnSOD水平,从而提高其抗氧化能力[26]。 Nmnat3的过表达有效地提高了多种小鼠组织中的NAD+,从而显著抑制ROS的生成,并防止与衰老相关的胰岛素抵抗[27]。NAD+还通过调节sirtuins和PARPs来调节细胞衰老过程中的氧化应激。NAD+依赖的SIRT1在氧化应激反应中显著上调,保护心脏免受氧化损伤,有助于延缓衰老[28]。
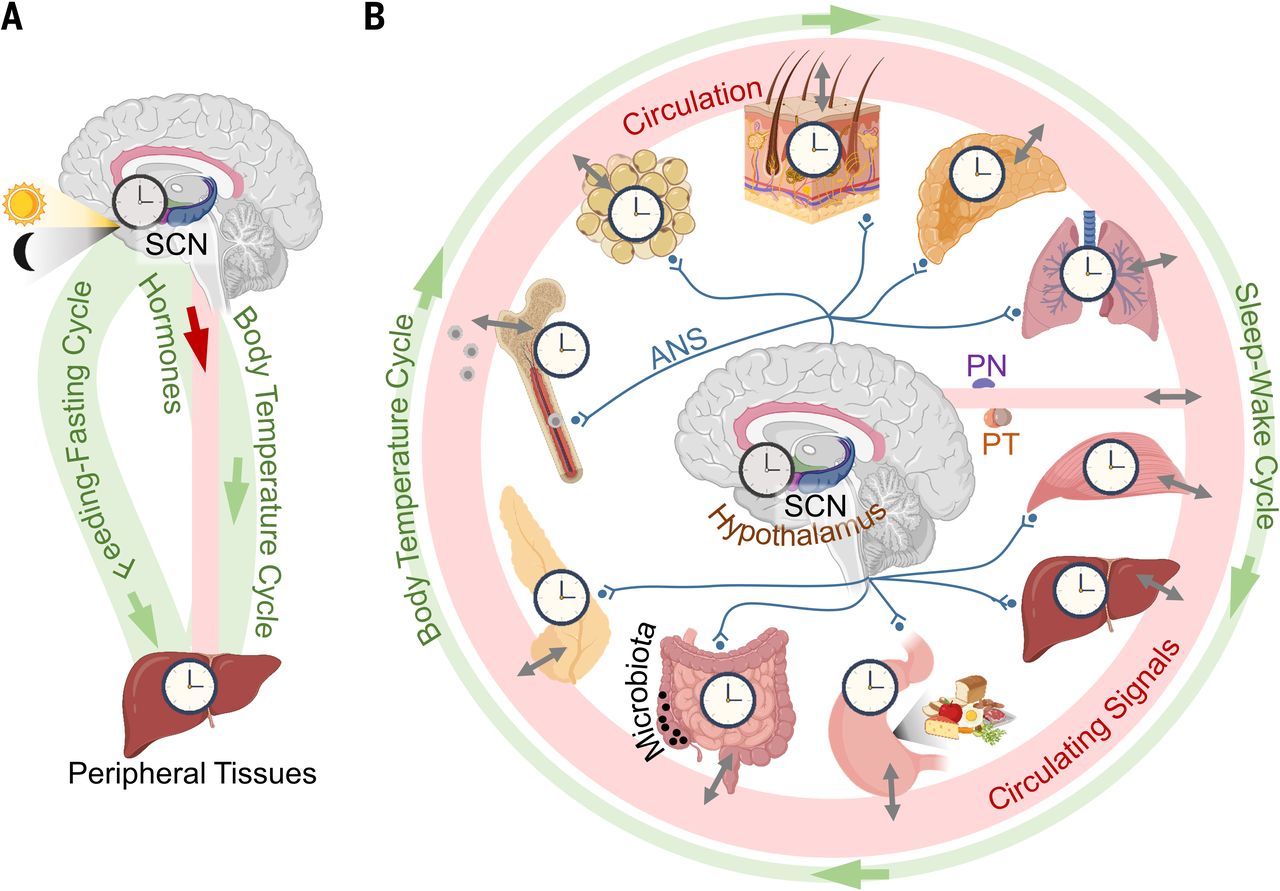
NAD+缺乏与衰老过程中的生物钟紊乱相关。除了减轻氧化损伤外,NAD+还可以通过驱动昼夜节律来延长寿命。生物钟是将代谢与外源性和内源性因素联系起来的内部计时器机制,它的失调与加速衰老有关[29]。在衰老过程中,昼夜节律蛋白将NAD+代谢与昼夜节律生物钟机制联系起来。SIRT1通过有节律地去乙酰化Bmal1或Per2诱导核心时钟基因(如Cry1、Per2、Rorγ和Bmal1)的昼夜节律转录[30,31]。SIRT1还调节昼夜节律启动子中H3 Lys9/Lys14的时钟介导的染色质重塑,以控制昼夜节律[31]。
在老年小鼠的中枢神经系统中,SIRT1水平显著降低,导致BMAL1等昼夜节律蛋白的降低[32]。自主肝时钟诱导NAD+挽救通路部分恢复NAD+振荡,即使没有其他时钟的输入,也能驱动肝脏中的SIRT1昼夜节律功能[33]。因此,NAD+依赖性SIRT1能够调节中枢昼夜节律功能随着衰老而下降。
衰老相关的核NAD下降通过SIRT1-HIF-1α-c-Myc通路抑制线粒体编码的基因,而提高NAD水平则以SIRT1依赖性方式挽救老年小鼠的线粒体功能[34]。
NAMPT在衰老人SMCs中的表达增加通过延缓衰老和增强对氧化应激的抵抗力来延长寿命[35]。
补充NAD前体NR和NMN,可提高NAD水平,通过激活小鼠体内的 SIRT1 来维持线粒体和代谢功能,从而延长小鼠的寿命[34,26,36]。
[1] Imai, S. & Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 24, 464–471 (2014).
[2] Powell, R. D. et al. Resveratrol attenuates hypoxic injury in a primary hepatocyte model of hemorrhagic shock and resuscitation. J. Trauma Acute Care Surg. 76,409–417 (2014).
[3] Aguirre-Rueda, D. et al. Astrocytes protect neurons from Aβ1-42 peptideinduced neurotoxicity increasing TFAM and PGC-1 and decreasing PPAR-γ and SIRT-1. Int. J. Med. Sci. 12, 48–56 (2015).
[4] Busch, F., Mobasheri, A., Shayan, P., Stahlmann, R. & Shakibaei, M. Sirt-1 is required for the inhibition of apoptosis and inflammatory responses in human tenocytes. J. Biol. Chem. 287, 25770–25781 (2012).
[5] Braidy, N. et al. Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PloS ONE 6, e19194 (2011).
[6] Zhou, C. C. et al. Hepatic NAD(+) deficiency as a therapeutic target for nonalcoholic fatty liver disease in ageing. Br. J. Pharmacol. 173, 2352–2368 (2016).
[7] Clement, J., Wong, M., Poljak, A., Sachdev, P. & Braidy, N. The plasma NAD(+) metabolome is dysregulated in “normal” aging. Rejuvenation Res. 22, 121–130(2019).
[8] Mouchiroud, L. et al. The NAD(+)/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 154, 430–441 (2013).
[9] Zhang, H. et al. NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 352, 1436–1443 (2016)
[10] Yoshino, J., Mills, K. F., Yoon, M. J. & Imai, S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 14, 528–536 (2011).
[11] Stein, L. R. & Imai, S. Specific ablation of Nampt in adult neural stem cells recapitulates their functional defects during aging. EMBO J. 33, 1321–1340 (2014).
[12] Camacho-Pereira, J. et al. CD38 dictates age-related NAD decline and mitochondrial dysfunction through an SIRT3-dependent mechanism. Cell Metab. 23,1127–1139 (2016).
[13] Kanfi, Y. et al. The sirtuin SIRT6 regulates lifespan in male mice. Nature 483,218–221 (2012).
[14] Scheibye-Knudsen, M. et al. A high-fat diet and NAD(+) activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab. 20, 840–855 (2014).
[15] Gomes, A. P. et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 155, 1624–1638 (2013).
[16] Peek, C. B. et al. Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science 342, 1243417 (2013).
[17] Venkataraman, K., Khurana, S. & Tai, T. C. Oxidative stress in aging-matters of the heart and mind. Int. J. Mol. Sci.14, 17897–17925 (2013).
[18] Barnham, K. J., Masters, C. L. & Bush, A. I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 3, 205–214 (2004).
[19] Jeong, Y. J. et al. Impact of long-term RF-EMF on oxidative stress and neuroinflammation in aging brains of C57BL/6 mice. Int. J. Mol. Sci. 19 (2018).
[20] Sun, M. et al. The need to incorporate aged animals into the preclinical modeling of neurological conditions. Neurosci. Biobehav. Rev. 109, 114–128 (2020).
[21] Royce, G. H., Brown-Borg, H. M. & Deepa, S. S. The potential role of necroptosis in inflammaging and aging. GeroScience 41, 795–811 (2019).
[22] Yang, D. et al. Pro-inflammatory cytokines increase reactive oxygen species through mitochondria and NADPH oxidase in cultured RPE cells. Exp Eye Res. 85,462–472 (2007).
[23] Fan, L. M. et al. Nox2 contributes to age-related oxidative damage to neurons and the cerebral vasculature. J. Clin. Investig. 129, 3374–3386 (2019).
[24] Stefanatos, R. & Sanz, A. The role of mitochondrial ROS in the aging brain. FEBS Lett. 592, 743–758 (2018).
[25] Li, S. Y. et al. Aging induces cardiac diastolic dysfunction, oxidative stress,accumulation of advanced glycation endproducts and protein modification.Aging Cell 4, 57–64 (2005).
[26] de Picciotto, N. E. et al. Nicotinamide mononucleotide supplementation reverses vascular dysfunction and oxidative stress with aging in mice. Aging Cell 15,522–530 (2016).
[27] Gulshan, M. et al. Overexpression of Nmnat3 efficiently increases NAD and NGD levels and ameliorates age-associated insulin resistance. Aging Cell 17, e12798 (2018).
[28] Alcendor, R. R. et al. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ. Res. 100, 1512–1521 (2007).
[29] Orozco-Solis, R. & Sassone-Corsi, P. Circadian clock: linking epigenetics to aging.Curr. Opin. Genet. Dev. 26, 66–72 (2014).
[30] Asher, G. et al. SIRT1 regulates circadian clock gene expression through PER2 deacetylation. Cell 134, 317–328 (2008).
[31] Nakahata, Y. et al. The NAD+-dependent deacetylase SIRT1 modulates CLOCKmediated chromatin remodeling and circadian control. Cell 134, 329–340 (2008).
[32] Chang, H. C. & Guarente, L. SIRT1 mediates central circadian control in the SCN by a mechanism that decays with aging. Cell 153, 1448–1460 (2013).
[33] Koronowski, K. B. et al. Defining the independence of the liver circadian clock.Cell 177, 1448–1462 e1414 (2019).
[34] Gomes AP, et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155:1624–1638. doi: 10.1016/j.cell.2013.11.037.
[35] van der Veer E, et al. Extension of human cell lifespan by nicotinamide phosphoribosyltransferase. J. Biol. Chem. 2007;282:10841–10845. doi: 10.1074/jbc.C700018200.
[36] Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14:528–536. doi: 10.1016/j.cmet.2011.08.014.

