
近年来随着生物技术的不断发展,出现了许多克隆新基因的方法和手段,如图谱克隆技术、转座子标签技术、mRNA差异显示技术、基因组减法技术以及cDNA文库筛选技术等。但上述方法大多具有实验周期长、技术步骤烦琐且工作量大等特点逐渐满足不了科研的需求,因此,RACE技术应运而生,接下来,就让小V带大家去了解一下RACE技术吧!~
你在做实验中还遇到过什么问题?留言回复点赞最高的同学可获得激光翻页笔一支。

RACE(rapid amplification of cDNA end)也叫cDNA末端快速扩增技术,是已知部分cDNA序列,基于逆转录和PCR快速克隆得到完整的cDNA的3'或5'末端的分子生物学技术。主要类型有3'RACE和5'RACE。

RACE 原理
(Vazyme#RA101)
Cap-switching 5'RACE原理
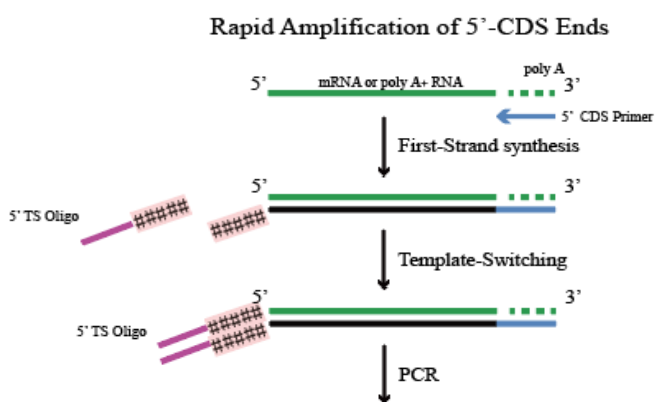
1.5' CDS Primer 识别RNA3'端poly A尾结构进行一链cDNA合成(若RNA无poly A尾,则可使用 5' Random Primer代替5'CDS Primer);
2.逆转录进行到mRNA 5'端时,逆转录酶的末端转移酶活性会在cDNA 3'末端加上d(C);
3.5' TS Oligo接头序列的3'末端带有polyG,可与cDNA末端的d(C)序列结合;
4.逆转录酶继续以5' TS Oligo为模板合成cDNA,完成接头转化;
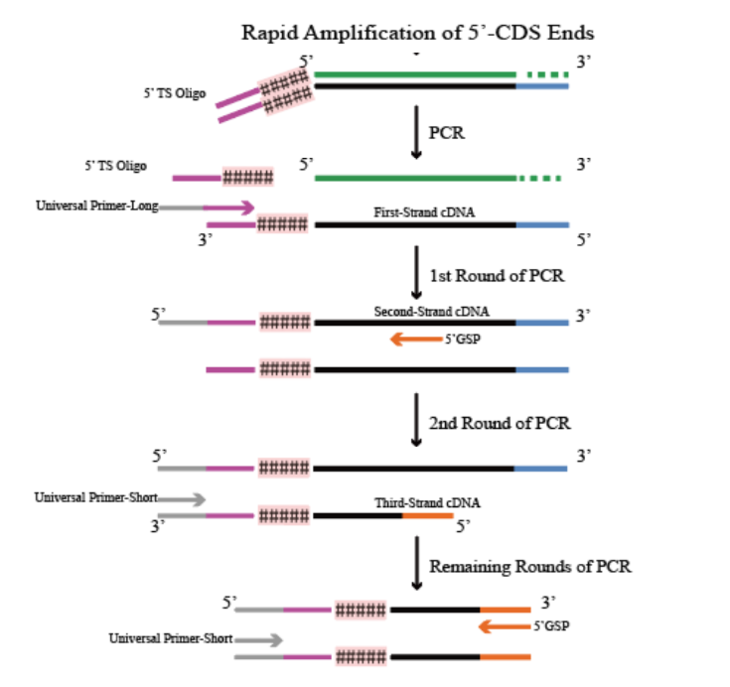
cDNA末端的快速扩增
5.UP - Long 带有通用引物序列和接头序列,与第一链cDNA 的3'端结合,合成第二链 cDNA ;
6.5'GSP 引物以第二链cDNA为模板合成第三链 cDNA;
7.UP - Short 为通用引物序列,与第三链 cDNA 的 3'端结合,合成互补双链 DNA;
8.UP - Short 与 5'GSP 引物以双链 DNA 为模板进行 PCR扩增。
经典3'RACE原理
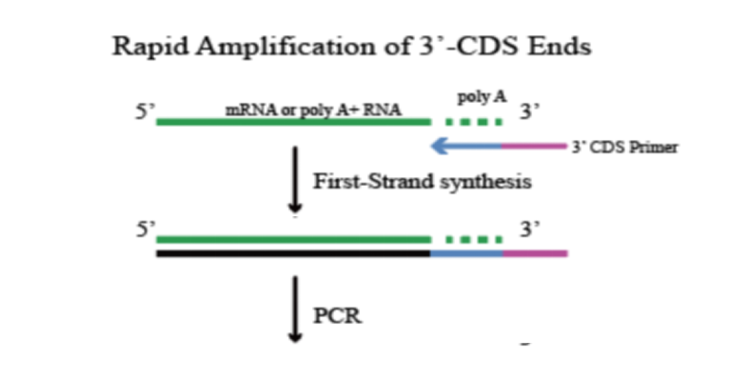
第一链cDNA的合成
1.3'CDS Primer 的5'端带有一段接头序列;
2.3'CDS Primer 识别 RNA 的3'端 poly A尾结构,逆转录合成一链cDNA(若RNA无poly A尾,则需进行加A处理);
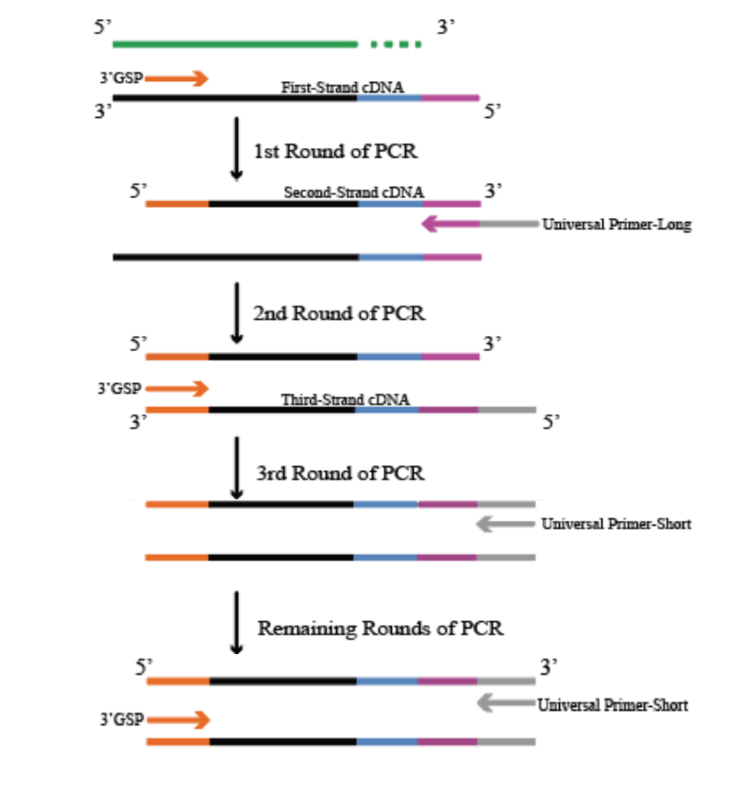
cDNA末端的快速扩增
3.3'GSP 以第一链 cDNA 为模板合成第二链 cDNA;
4.UP - Long 带有与3'CDS Primer 相同的接头序列,识别第二链cDNA的3'端,合成第三链 cDNA;
5.3'GSP 以第三链 cDNA 为模板合成双链 DNA;
6.3'GSP 与 UP - Short 以双链 DNA为模板进行PCR扩增。
引物设计的优劣对于做RACE是至关重要的,如果引物的特异性不好,就会给实验带来许多麻烦,甚至扩增不到我们实验预想的目的产物,接下来小v就带大家看看RACE实验中需要用到的引物设计原则吧!
5'/3' GSP(基因特异性引物)设计原则 :
引物长度:23 - 28个核苷酸,建议不超过30个核苷酸;GC含量:50% - 70%;
Tm值:Tm≥65°C,Tm>70°C使用Touch Down PCR有助于提高产物特异性;对于较长片段(>10 kb)或丰度较低的待测转录本,GSP引物设计尽量靠近cDNA的末端(≤3 kb);针对同一待测转录本可设计多条GSP,进行RACE扩增以提高成功率。
NGSP(巢式基因特异性引物)设计原则(可选):
1.引物长度、GC含量、Tm值:同GSP设计原则;位置:NGSP须设计在GSP的3'端,建议NGSP的5'末端与GSP的3'末端有5 - 15 nt的重叠,有助于提高二轮巢式扩增时的特异性。
2.如果上一轮 PCR 没有理想的特异产物(如无扩增产物、产物较弱、产物弥散),或者产物非特异条带过多、基因表达丰度低,巢氏PCR与RACE结合可以提高克隆的准确度,巢式引物结合在上一轮PCR产物的内部,进行PCR扩增可设计多轮巢式引物进行巢式PCR扩增。
友情提醒:巢式PCR可以提高扩增倍数,降低了非特异性反应连续放大进行的可能性,提高了实验的成功率。
1) 扩增较长片段有什么建议?
➣ 建议扩增片段尽量不超过3 kb,靠近已知序列末端;
➣ 设置温度梯度摸索最佳退火温度;
➣ 检测样本RNA是否降解;
➣ 扩增较长片段时应该适当延长变性时间;
➣ GSP引物设计需要保证较好的特异性;
2) 进行cDNA末端快速扩增时没有明显产物条带?
➣ RNA提取物中无目的转录本或者目的转录本表达丰度低,可以设计已知转录本区域的qPCR或PCR引物进行目的产物检测;检测后发现表达丰度低,可以增加RNA模板投入量;
➣ RNA降解:进行试验前检测RNA的完整度,可以通过RNA琼脂糖凝胶电泳观察RNA的完整度;
➣ 设计Nested GSP,进行多轮巢式PCR(一般不超过3轮),富集产物片段同时保证了扩增产物特异性;
➣ 待扩增目的片段过长:提高PCR延伸时间,GSP设计时尽可能放在序列末端;
3) PCR出现明显杂带?
➣ 目的基因属于多基因家族,可以进行数据库对比,针对特异性区段序列设计GSP;
➣ 可能是RNA前体具有可变剪接形式,可以进行数据库对比,针对特异性区段序列设计GSP;
➣ 扩增非特异性:设计额外的NGSP,进行多轮巢式PCR,将目的产物进行胶回收,再次进行RCP扩增。
4) 5' RACE成功,3' RACE失败是什么原因?3' RACE成功,5' RACE失败是什么原因?
➣ 如果5'RACE能做出来,3' RACE做不出来,大概率是GSP引物的问题;如果3'RACE成功,5' RACE失败,可能与RNA完整度有关。
5) RNA不含poly A尾,加A处理后做了5' RACE 没有出现目的条带?
➣ 逆转录后设计已知序列的qPCR引物或PCR引物,扩增逆转后的cDNA,如果没有扩增说明没有cDNA或含量较低,建议使用随机引物进行逆转,同时选择多条GSP进行PCR;如果有扩增产物,说明逆转录没有问题,可以进行巢式PCR富集特异产物。
6) 引物设计结果为空白是什么原因?
➣ 引物设计结果为空白,可能是没有符合要求的引物序列,可以排查一下引物GC含量,GC含量太低或太高都会产生影响。
7) 5' RACE测序结果与软件比对结果相比多出3个G碱基?
➣ 测序结果多出3个G碱基是RACE过程中加上去的,对实验结果不会产生影响,软件比对时把多出的碱基删除为真正的5'端序列。
8) 克隆产物测序结果不同,如何判断哪个是全长序列?
➣ 转录本本身可能存在多种可变剪接形式,表明两个序列都是正确的。尽量在实验中尽可能地增加克隆数测序,减少实验中的误差。
好啦~,今天的知识点小v就介绍到这里,如果在实验过程中有什么不懂的,可以随时来询问小v,或者直接联系身边的诺唯赞人,我们将竭尽全力为大家提供优质的服务和可靠的产品。
留言回复点赞最高的同学:可获得激光翻页笔一支。
本活动于 2022 年 1 月 17 日 17 点截止,获奖后会在公众号后台通知,欢迎留言,最后,希望大家做实验都能做出完美的结果。

