


商品详情
 书名:遗传性听力损失及其综合征
书名:遗传性听力损失及其综合征
定价:398.0
ISBN:9787117307925
作者:王秋菊,杨仕明,赵立东,韩东一
版次:1
内容简介
纵本书已经成为遗传性听力损失及其临床表现方面必备案头书和供查阅“大词典”。第3版在前两版基础上,对流行病学特点、胚胎学、非综合征型和综合征型听力损失的章节都进行了更新,特别对是有关分子机制方面的海量的新信息给予了特别关注。本版共分为18章,每个章节都由该领域内的知名专家来撰写,各个章节详细介绍了与遗传性听力损失相关的基础知识,各种综合征在全身各系统(视觉系统、代谢系统、心血管系统、神经系统、肌肉骨骼系统、内分泌系统等)的临床表现、发病机制、实验室检查、影像学检查、病理学检查、遗传特征、分子遗传学研究、诊断、小结等,并结合了基因组学和遗传学的新技术、新进展,更具实用性和前沿性。这一卷是写给执业临床医师的,对于临床医师、听力学家和其他为耳聋患者及其家庭服务的专家来说,这本书都是极好的案头参考,也可以作为临床培训计划和进行听觉科学研究人员的教科书。
编辑推荐
一书囊括/ 涵盖一切常见的、经典的、罕见的遗传性听力损失相关综合征
权威可信/ 原著为国际经典,译著由国内知名专家审译把关
更便查阅/ 在原著目录基础上增加提取基因座和各综合征
主译简介
王秋菊:主任医师,教授,博士研究生导师,解放军总医院耳鼻咽喉头颈外科医学部耳鼻咽喉内科主任、全军耳鼻咽喉研究所所长。
杨仕明:主任医师,教授,博士研究生导师,解放军总医院耳鼻咽喉头颈外科医学部主任、国家耳鼻咽喉疾病临床研究中心主任。
赵立东:副研究员,硕士研究生导师,解放军总医院耳鼻咽喉头颈外科医学部耳鼻咽喉内科、全军耳鼻咽喉研究所。
韩东一 :主任医师,教授,博士研究生导师,解放军总医院耳鼻咽喉头颈外科医学部、全军耳鼻咽喉研究所名誉所长。
内 封

审校专家及其单位

译者及其单位


第3版译者序

原著前言

目 录











样章试读
歌舞伎综合征
Kabuki 综合征
Kabuki syndrome
1981 年,Niikawa 等 [11]和 Kuroki 等[8]同时独立报道了一种在日本儿童中发现的综合征,主要表现为轻度至中度的智力障碍,出生后生长发育迟缓,骨骼异常以及一张十分怪异的类似歌舞伎面具样的脸。这种病称为 Kabuki 综合征(歌舞伎综合征),目前已经有世界各地不同种族的病例报道 [1]。
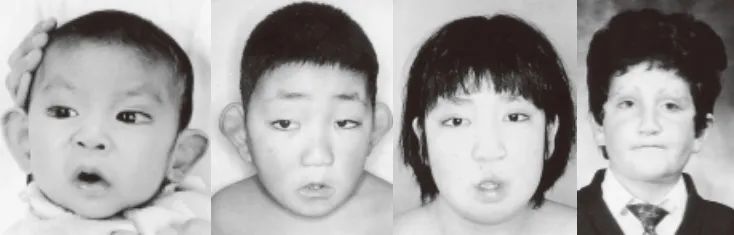
颅面部表现:
睑裂长,下睑外 1/3 外翻。拱起的眉毛两侧变淡,睫毛长而浓密。最常见的眼部特征包括蓝巩膜、上睑下垂和斜视。鼻部表现为鼻宽且鼻尖扁平(图 13-1)。牙齿小、稀疏,一些恒牙缺失。70% 患者有唇裂和 / 或腭裂、黏膜下腭裂,或硬腭高拱,也有报道下唇凹陷 [16]。
肌肉骨骼系统:
常见的畸形包括身材矮小,短和 /或弯曲的第五指,脊柱侧弯(25%)以及各种肋骨和脊椎畸形(20%~30%)。75% 的患者有关节的超活动性,其中先天性髋关节脱位占20%,其他关节如肩、膝关节异常较少见 [1]。
心血管系统:
Kabuki 综合征中约一半的病例报道有心脏缺陷[4]。最常见的是房间隔缺损、室间隔缺损和主动脉缩窄[5,16],也有报道二叶式主动脉瓣。主动脉缩窄及二叶式主动脉瓣可导致全身血流量阻塞(左心阻塞性缺陷)。其他报道 Kabuki 综合征的左心阻塞性缺陷包括主动脉瓣狭窄、主动脉瓣下狭窄、二尖瓣狭窄、降落伞式二尖瓣和左心发育不良。整体上看,Kabuki 综合征出现左心阻塞性缺陷与正常人群比较具有统计学意义。这些缺陷的高频出现与 Turner 综合征相似。但是,Kabuki 综合征心脏异常的种类与Turner 综合征相比更为广泛,病例报道的其他缺陷有肺动脉瓣狭窄、右心室双出口、法洛四联症、大血管转位、静脉回流异常症[2,4,5,16]。
皮肤系统:
90% 患者存在持续性胎儿样指尖[1]。
听觉系统:
突出的大耳郭上常常有一对大耳垂。对耳轮往往发育不全,呈杯状耳外观,约 20% 患者有耳前凹。患者儿童期发生中耳炎的比例很高[1]。尽管感音神经性听力损失极少见[3],但仍有 24%~82% 病例出现传导性、感音神经性或混合性听力损失[1,6,13]。可出现听骨链畸形或固定[15],也有 Mondini 畸形的报道[6]。
前庭系统:
少数测试过前庭功能的 Kabuki综合征患者表现是正常的[3]。
遗传学:
迄今报道的 Kabuki 综合征患者绝大多数没有家族史,无性别差异。有少数病例报道存在家族史[16]。
分子生物学研究:
具有典型表型患者中 90%能检测到 MLL2 基因杂合突变[10],而在大规模患者人群中检测发现 60%~75% 存在 MLL2 基因杂合突变[10,12]。在 2 个家族性病例中,父母和孩子都发现了 MLL2 基因突变[10]。MLL2 是一种组蛋白甲基转移酶,作为转录激活因子,参与细胞黏附、运动和生长[7]。最近 3 例 Kabuki 综合征被发现有 KDM6A 基因内缺失或全部的微缺失,这个基因表达一个组蛋白去甲基化酶,与 MLL2 基因相互作用[9]。但还有一些患者的基因研究中并未发现这两个基因的任何突变或缺失 / 重复,表明很可能存在遗传异质性[14]。
小 结:
特点包括:
①绝大多数为散发病例,偶有常染色体显性遗传;
②独特的面部特征,包括睑裂狭长和招风耳;
③出生后生长发育迟缓;
④轻、中度智力障碍;
⑤心脏畸形;
⑥频发中耳炎;
⑦听力损失可为传导性、感音神经性或混合性。
- 人卫智慧服务商城 (微信公众号认证)
- 扫描二维码,访问我们的微信店铺
- 随时随地的购物、客服咨询、查询订单和物流...









